Why Your AI Is Reading the Wrong Half of the Protein
When AlphaFold assigns a region of a protein a confidence score below 50, most downstream pipelines treat that as a problem to be solved. The structure looks unreliable. The protein looks fragile. Some target prioritization systems even penalize candidates for it.
That interpretation is often a category error — not because the AI is wrong, but because the conclusion drawn from it is.
The issue is not that AlphaFold is mispredicting structure. It is that many downstream interpretations ignore decades of structural biology showing that lack of stable structure can itself be functional. This shift is formalized in what researchers now describe as the "disorder–function paradigm," where biological activity can arise from flexibility rather than from fixed form.
The short version: a large fraction of the human proteome is not meant to adopt a stable three-dimensional structure. Roughly one third of residues exist in disordered regions, and more than half of human proteins contain at least one such segment. These regions are not rare edge cases — they are widespread and central to protein function, often existing as dynamic ensembles of interconverting conformations rather than a single fold.
When a model assigns low confidence to these regions, it is usually reporting an accurate physical reality. The mistake is treating that reality as a defect rather than a feature.
In many cases, the disorder is the function.
The first principle — and where the AIs are genuinely strong
Proteins fold because of water. Hydrophobic amino acids — valine, leucine, phenylalanine — drive collapse into a tightly packed core to minimize disruption of the surrounding solvent. This hydrophobic collapse is one of the closest things structural biology has to a law.
This is the domain where AlphaFold and OpenFold3 excel:
- well-folded cores
- stable helices and sheets
- tightly packed interiors
If your analysis focuses on this layer alone, it is often directionally correct. It just isn't the whole system.
Functional disorder: the part that looks wrong but isn't
For decades, biology operated under the assumption that structure determines function. That is now known to be incomplete.
Intrinsically disordered proteins do not settle into a single shape. Instead, they exist as ensembles of conformations, with function emerging from their dynamic sampling behavior rather than a fixed structure.
These regions:
- enable transcription factors to bind multiple partners
- provide docking platforms in signaling pathways
- drive liquid–liquid phase separation
None of this works if the region is rigid.
So when AlphaFold assigns a low confidence score to these segments, it is not failing. It is detecting the absence of a stable structure. Early analyses of AlphaFold outputs showed that many low-confidence regions correspond to proteins already known to be intrinsically disordered — regions that "have no fixed structure… and that's valuable," as researchers noted when interpreting these predictions.
Follow-on studies have emphasized that caution is required when applying structure-based reasoning to these regions, as treating them like folded domains can misrepresent their functional role.
A scoring system that penalizes disorder is therefore systematically biased against proteins doing some of the most regulated and therapeutically relevant work in the cell.
Allostery: the half of biology static structures cannot show
A protein's active site is only part of the story.
Allostery allows binding at one location to alter behavior at another. This is fundamental to:
- feedback regulation
- signaling cascades
- drug specificity
AlphaFold predicts a dominant conformation. Allostery depends on transitions between multiple conformations.
This highlights a broader limitation: many proteins — especially those involved in signaling — are better described as dynamic ensembles. Any single predicted structure captures only one snapshot of a fundamentally fluctuating system.
That means analyses built purely on static structure are not wrong. They are incomplete.
Folding on the fly
Proteins begin folding while they are still being synthesized. As the ribosome produces the chain, earlier segments start adopting structure before later segments even exist.
This process — cotranslational folding — means:
- folding pathways are constrained
- intermediate states matter
- final conformations depend on cellular context
A predicted structure represents a plausible endpoint, not necessarily the biologically realized pathway. Any model that assumes otherwise is simplifying a system that is inherently dynamic.
The post-translational layer
A newly synthesized protein is not finished. It is modified.
Post-translational modification — phosphorylation, glycosylation, ubiquitination, methylation, acetylation — controls:
- activity
- localization
- stability
- interaction networks
These modifications frequently occur in disordered regions, where enzymes have physical access to target residues.
This creates a critical intersection:
disorder + modification = regulatory control
And it is almost entirely invisible to structure-only models.
Where current pipelines go wrong
The issue is not AlphaFold itself. The issue is how its outputs are used.
Many target prioritization systems implicitly assume:
- high structural confidence = good
- low structural confidence = risk
That collapses two fundamentally different phenomena:
- failure to fold (true instability)
- designed disorder (functional flexibility)
Treating them as the same leads to systematic bias.
A better mental model
Instead of viewing proteins as static objects, it helps to think in three layers:
- Structured core (high confidence) — catalytic activity, stable scaffolding
- Disordered regions (low confidence) — signaling, interaction flexibility
- Regulatory layer (post-translational modification) — control and timing
Function emerges from the interaction of all three — not from structure alone.
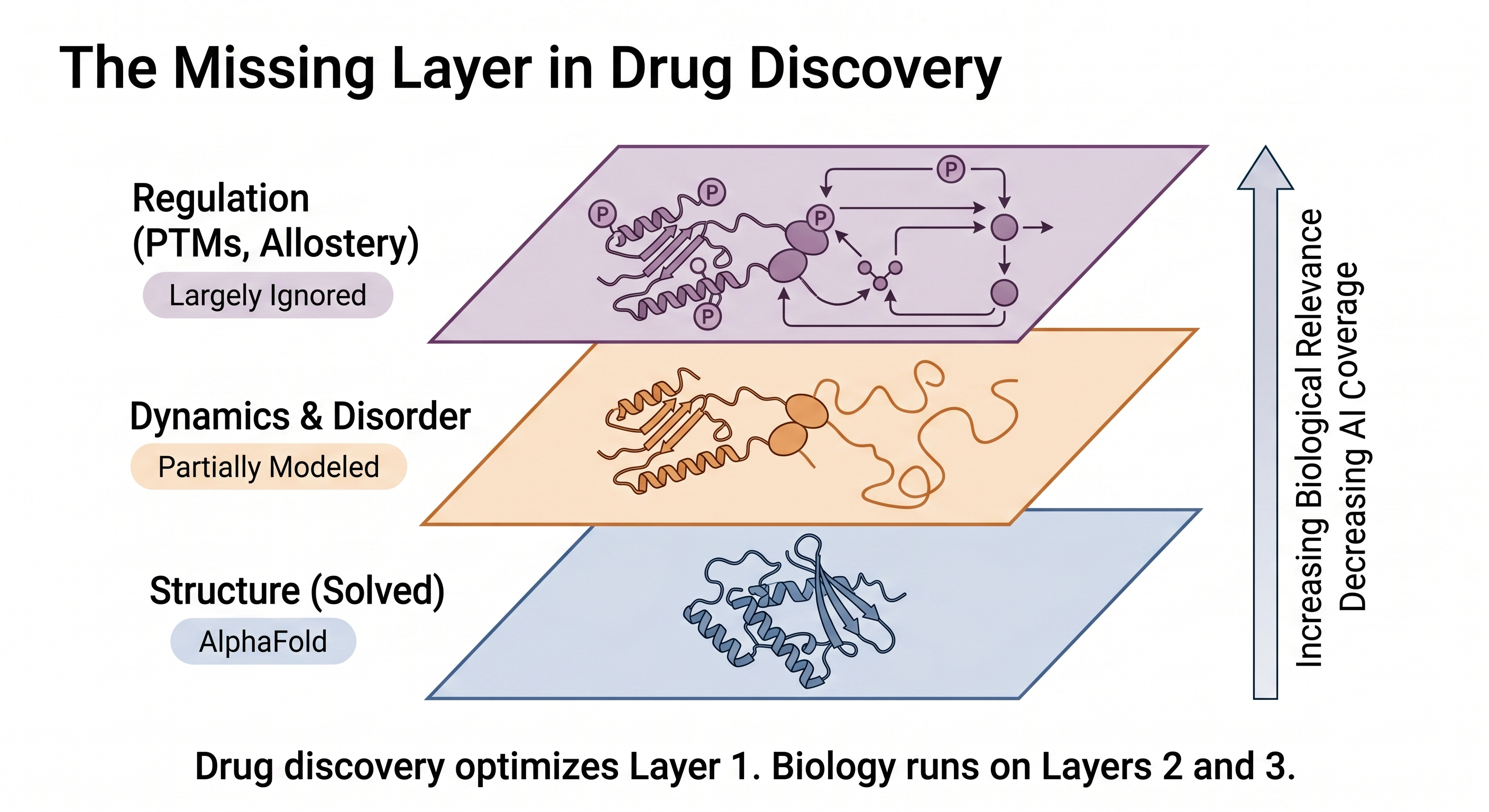
Drug discovery has spent the last few years optimizing Layer 1. Biology runs on Layers 2 and 3.
Why this matters in real targets
Different classes of proteins emphasize different layers.
- Highly structured targets like PCSK9 or SGLT2 align well with traditional structure-based approaches.
- Regulatory proteins and transcription factors rely heavily on disorder and modification.
- Even clinically successful pathways, such as GLP-1 signaling (e.g., Ozempic), involve dynamics and regulatory layers that extend beyond a single static structure.
Treating all targets with a single structural lens misses these differences. The pathway layer — the cell-type-specific, time-resolved representation of how biology actually works — is where Layers 2 and 3 live in practice. We haven't built it yet.
A simple test for any AI-generated analysis
If you are evaluating a "druggability score" or "structural risk index" — yours or a vendor's — ask:
- Does it distinguish between instability and intrinsic disorder?
- Does it account for allostery and conformational dynamics?
- Does it incorporate post-translational state?
If the answer to any of these is no, you are looking at a partial model presented as a complete one. The 14 models NVIDIA AI Enterprise ships under Life Sciences are mostly Layer 1 — strong on structure, generation, and docking; conspicuously thin on the regulatory and dynamic layers. That isn't NVIDIA's failure. It's an industry-wide one.
The real opportunity
Static structure prediction is a major breakthrough — but it represents only one layer of the problem, and is largely solved for many single-chain proteins.
The next frontier is integration:
- disorder
- dynamics
- allostery
- post-translational modification
The companies that succeed will not just predict structure. They will model what proteins actually do inside living cells.
Because in many of the most important systems — metabolism, signaling, transcription — the critical biology is happening in the parts of the protein that current models see least clearly.
A low confidence score is not always bad news.
Sometimes, it is the protein telling you exactly where the action is.
Share this post
Tagged

